|

|
Van Lars Fischer, Südollnborger Platt van Ludgerd Lüske Vandaoge schaffet dei Computer hochpräzise Biller van Enzyme in Aktion. Aower dortau mössen dei Wätenschkupslüe eiers eis twei dörweg unnerscheidlicke Verfohren tausaomebringen. Dei Lohn: Ein wohrhaft einmaoliget Verbinnen tüschken Theorie un Praxis. Form un Funktion sünd in dei Chemie faste miteinanner tausaomeknüppet, bi einfach upbawte Moleküle kann man meist nao einen korten Blick up dei Strukturformel wäten, wo sick disse Stoff bi chemischke Reaktionen vehollt. Dei interessantesten Moleküle sünd aower väl komplexer. Bi dei finnt man tau'n Bispill wisse Biomoleküle, dei aal dei unnerscheidlicken Reaktionen utföhrt, dei eine lebennige Zelle bruket. Ein Menschk kann aal dei Reaktionen van sükke Proteine all lange nich mehr vörutseihn — dei Computer aower kann dat. Dorför, dat Computerprogramme vandaoge Form un Verhollen van Strukturen mit Duusende Atome binnen mit Verlaot beräken käönt, häbbet nu Arieh Warshel van dei University of Southern California, dei in Wien born Martin Karplus un dei in Pretoria born Michael Levitt den Nobelpries för Chemie krägen.
Den Anfang up dissen Weg möken dei Chemiker all in dei 1940er Johre, lange vördem et Computer für sükke Upgaoben geef. Sei äöwerleggden sich don, wekke Deile van ein Molekül sick elektrostaotischk antrecket off affstötet, un off einkelne Atome unner wisse Ümstände miteinanner tausaomestötet. Ut disse einfachen Gedanken entwickelden sick dei eiersten Modelle up dei Grundlaoge van dei potenzielle Energie van Atombülte un heele Moleküle. An sükke Methoden arbeiteden Warshel un Levitt in dei 1960er Johre tausaome mit den beduurlick 2002 storven Shneior Lifson. Warshel un Lifson publizeierden 1968 dat Consistent-Force-Field-Verfohren (CFF-Verfohren). Bi disse Methode werd mit väle Räkenformeln, dei dat Wesselwarken un dei Energiekurven dortau beschriewet, dei passen Form uck för komplexe Moleküle beräket. Molekülmodelle för Dröhnbüdels Dei naue Form van ein Molekül tau bestimmen, is ansick ein Optimierungsproblem: Man söch dei leegste potenzielle Energie van ein System ut väle aneinanner bunnen Atome. Tau'n Bispill hätt dei Räkenformel, dei dei Energie van eine chemischke Bindung beschriff, ein Minimum bi eine wisse Längte — annere Längten sünd in'n Vergliek ungünstiger. Is dei Bindung so tau kort off tau lang, optimeiert dat Kraftfeldodell so lange wieder, bit dei minnste Energie un dormit dei günstigste Bindungslängte funnen werd. Dormit man up disse Oort ein Molekül korrekt beräken kann, mäötet väle Wesselwarke upmaol otimeiert wern — tau dei Bindungen uck noch elektrostaotischke Kräfte tüschken uplaodte Moleküldeile off dei so neumten Van-der-Waals-Kräfte, dei upträet, wenn dei Elektronenhüllen van Moleküldeile sick anstötet.
Dat lutt nao einen heelen Barg Räkenarbeit — aower man kann den Upwand äöwerkieken, eierst recht in'n Vergliek mit dei quantenmechaonischken Methoden. Up dei einen Siete simuleiert man mit sükke klassischken Methoden heele Atome off uck Moleküldeile, wobi dei Objekte van dei Quantenmechaonik dei einkeln Elektronen un Atomkarne sünd. Up dei annere Siete is dei klassischke Physik dörweg einfacher — so eine potenzielle Energie lätt sick fix beräken, quantenmechaonischke Modellierungen dorgägen sünd ein heelet Stück komplizierter un bruket mehr Räkenupwand. Dorümme käönt dei Forschkers mit dei CFF-Methode uck groote Moleküle bewarken, för dei mit qantenmechaonischke Methoden Johrteihnte an Räkentied nödig wör — tau'n Bispill bi groote Biomoleküle so as bi dei Proteine. Dynamischke Modelle bruket Quantendynaomik Dat Problem dorbi is: Kraftfeldmethoden beräket Gliekgewichtsstrukturen. Un dei nähmt dei Moleküle blots denn in, wenn nix ehre Krings stört. Dormit sünd aale chemischken un physikaolischken Reaktionen utschlaoten. Wenn aower mit ein Molekül wat Interessantet passeiert, et tau'n Bispill eine Bindung mit ein anneret Molekül ingeiht, versägget disse Methoden. Quantenmechaonischke Operationen, dei parallel tau dei klassischken Modelle upkömen, sünd dorgägen dynaomischk. Dei wiest ein Bild van dei lebennige Chemie, dei sick wietaff van dat Gliekgewicht affspält, in dat dei Moleküle anreget werd, entstaoht un vergaoht. Mit sükke Methoden faotet man blots dei chemischken Reaktionen, dei för dei Funktionen van Biomoleküle ein Bedüden häbbet, un beläwet Enzyme in Aktion.
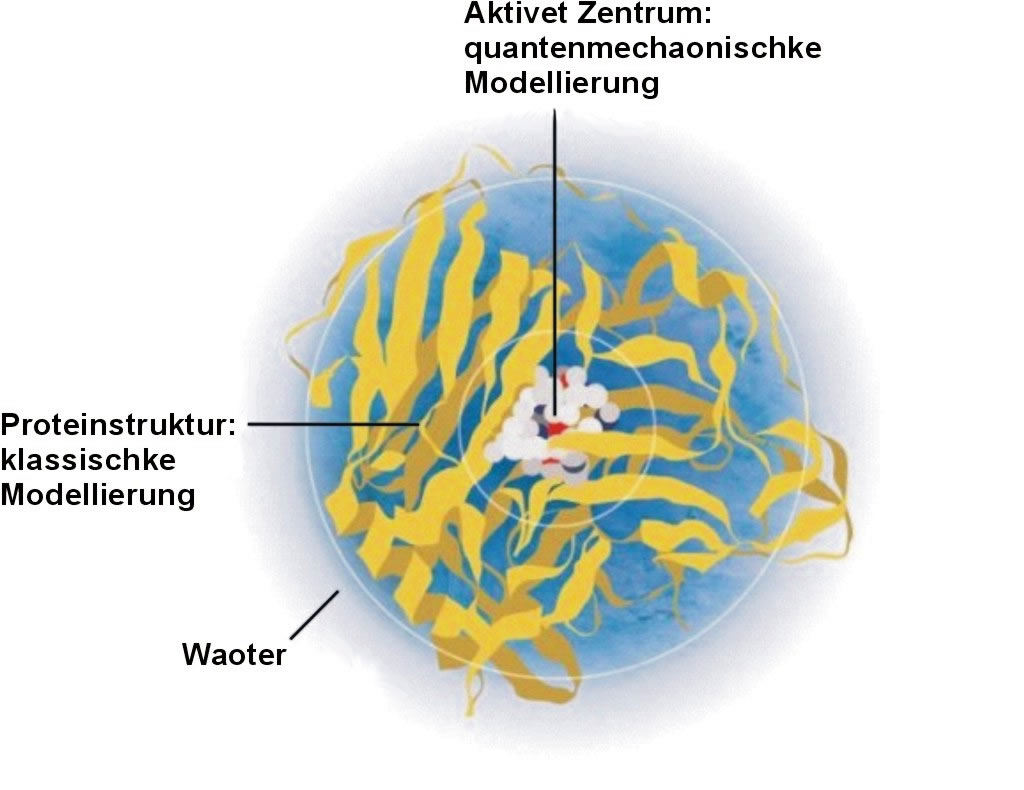
In Wohrheit deit man dat aower nich. Nämlick dorbi verbrukt dei Quantenmechaonik för Molekülsimulationen so väl Räkenkraft, dat et nich angeiht, dat man dei aktuellen Computer ein koplettet Protein up disse Ort dörräken lätt. Reaktionen tüschken lüttke orgaonischke Moleküle kunn man mit disse Methoden eintied bewarken — man jüst bi dei Biochemie mit ehre groote Bandbredde an Reaktionen, Produkte un Katalysatoren haren dei Computermodellierer schiens för immer kiene Chance. Dei Lösung van dat Problem lutt achternao recht einfach un ligg dune bi: Man arbeitet up dei verschiedenen Gröttenskaolen mit unnerscheidlicke Methoden. Dat komplette Molekül butenümmetau kann man mit dei klassischken Physik un so mit minne Räkenupwand beschriewen. Un in dat aktive Zentrum, wo sick dei eigentlicke Reaktion affspält, settet man dei dynaomischke Quantensimulation in. Dei eiersten Aktionen in disse Richtung mök man nich an komplexe Biomoleküle, sünners an dorgägen orig einfache aromatischke un unsatte Kohlenwaoterstoffe, woan Karplus un Warshel in dei 1970er Johre warkden. Don versöchden sei, dei Schwingungs- un Elektronenspektren van disse Moleküle tau simuleiern. Dortau brukden sei eiers eis dei Bindungslängten un -winkels in't Molekül, dei sei mit dei klassischken Kraftfeldmethoden bestimmden. Hybride Molekülmodelle Baobenskoop kaomt in disse Stoffe aower uck dei Elektronen van dei Dubbelbindungen vör, dei sick frei äöwer groote Rüme bewägen käönt un dei ehre naue quantenmechaonischke Eigenschkup ein Bedüden häbbet för dei vörutseihn Absorptionsspektren. Disse soneumten konjugeierten Elektronen modelleierden Karplus un Warshel mit quantenmechaonischke Methoden. Disse hybride Ansatz bröchde den Erfolg, un dat, wo doch dei Forschkers twei heel verschiedene physikaolischke Modelle tausaomeknüppden. In disse freuhe Arbeit funktionierde dat uck dorümme, wiel dei unnersöchten Moleküle plaon un stief sünd. Dat "klassischke" un dat "quantenmechaonischke" Elektronensystem sünd up disse Ort all rüümlick updeilt un dei doch so unnerscheidlicken Methoden kriegt sick nich in dei Klatten. All ein poor Johre Läöter brobeierde Warshel ut, ditmaol tausaome mit Levitt, off man disse besünnern Raohmenbedingungen woll weglaoten kunn. Dortao nöhmen disse Forschers einkomplexet Biololekül: Dat Lysozym, ein Enzym, dat Zuckerkäen upkielt. Dormit man disse Raktion Simuleiern kann, mutt man blots dei würklich bi dat Upkielen mitmaoken Strukturen quantenmechaonischk behanneln — dei groote Rest van dat Enzym werd wiederhen klassischk modelleiert. Grenzverkehr tüschken Quanten-Land un Klassik-Land Hier sünd aower beide Deile pass miteinner verknüppet und bewarket sick gägensietig. Dei Upgaobe hiertau hätt wat tau daun mit ein Problem ut dei Wirtschkup: Wo organiseiert man den Hannel tüschken twei Länder mit unnerscheidlicke Währungen? Man mutt ein System finnen, dat man an dei Grenze eine Währung mit Verlaot un mit bekannte Kurse gägenenanner ümmetuschken kann; dei Währung, mit dei Atome un Moleküle hannelt, is Energie. Aohne up dei grundsätzlicken Unnerscheide tüschken dei Methoden tau achten, fünnen Levitt un Warshel Verfohren, den Transfer van Energie äöwer dei Grenzen weg tüschken dat "Klassik-Land" un dat "Quanten-Land" pass tau beschriewen, uck för dat Wesselwark tüschken Moleküldeile ut beide Regionen. Dör dissen Erfolg kunn man grundsätzlick, in den Raohmen van ein klassischket Molekülmodell, dynaomischke Reaktionen quantenchemischk beräken. Sietdem bruket man in dei Chemie faoken disse Möglichkeit.
Dei modernen produktiven Computermodelle, dei Karplus, Levitt un Warshel don utklamüsterden, läwert vandaoge so präzise Daoten, dat man dei aohne Probleme mit würklich mäten Werte ut dat Labor verglieken kann. So kann man wohrhaft mit disse Methoden ein grootet Problem in dei chemischken Forschkung achter sick laoten — dat dei Chemiker ehre Moleküle un Reaktionen nich direkt seihn käönt. Moderne Computersimulationen stellt nich blots vördem nich tau griepen Daoten her, dei schaffet uck beste Biller, dei eine heel neie Ort van chemischke Forsckung möchlick maoket: dei beiden, Moleküle un Reaktionen, in't Gesicht tau kieken. Mehr Information: Moleküle un ehr binnen liggen Wesselwark |
||
|
Originaolartikel hier 20.10.2013 |
 Chemie-Nobelpries
2013: Form und Funktion in'n Computer simuleiert
Chemie-Nobelpries
2013: Form und Funktion in'n Computer simuleiert